|
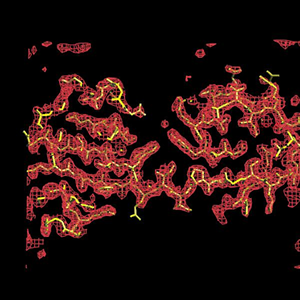
SOLVE/RESOLVE electron-density map and auto-built atomic model for initiation factor 5a at a resolution of 2.1A. Multiwavelength diffraction data courtesy of Tom Peat; refinement with Garib Murshudov's refmac5; graphics drawn with Alwyn Jones' O |
- SOLVE:
- Automated crystallographic structure solution for MIR, SAD, and MAD
- RESOLVE:
- Statistical density modification, local pattern matching, automated model-building, automated ligand-fitting, and prime-and-switch minimum bias phasing
Thomas C. Terwilliger -
(Tom Terwilliger) -
Los Alamos National Laboratory
Download SOLVE/RESOLVE website PDF (12 Mb) including the SOLVE/RESOLVE manual
Subscribe to the SOLVE / RESOLVE mailing list!
References for SOLVE and RESOLVE
SOLVE: Terwilliger, T.C. and J. Berendzen. (1999) "Automated MAD and MIR structure solution". Acta Crystallographica D55, 849-861.
RESOLVE: Terwilliger, T. C. (2000) "Maximum likelihood density modification," Acta Cryst. D56, 965-972.
RESOLVE model-building: Terwilliger, T. C. (2003) "Automated main-chain model building by template matching and iterative fragment extension" Acta Cryst. D59, 38-44.
If you like SOLVE/RESOLVE...try the new PHENIX software!
PHENIX can do everything SOLVE/RESOLVE can do, plus much more. It is a fully integrated system for solving structures by MIR/MAD/SAD (using SOLVE and HYSS) or molecular replacement (using Phaser), density modification (using RESOLVE) and iterative model-building and refinement (using RESOLVE and phenix.refine). Plus PHENIX has many terrific tools including Xtriage for analysis of datasets, elbow for generation of restraints, find_helices_strands for quick identification of secondary structure in a model, resolve_trace for multi-crystal averaging, and much more! You can run PHENIX from a GUI or from simple command-line scripts and parameters files and it is easy to modify runs you have already done to create new ones. If you want to run a SOLVE/RESOLVE script you can do it in PHENIX too with phenix.solve and phenix.resolve. It's all free for academics/non-profits at http://www.phenix-online.org .
Features of version 2.13 of SOLVE / RESOLVE
RESOLVE can now use multi-domain NCS in density modification. RESOLVE now allows you to specify up to 100 separate regions for applying NCS symmetry. Each one can have a separate set of NCS operators. The regions can be specified with PDB files.
Automated loop fitting RESOLVE now has a very powerful loop-fitting algorithm. You provide the residues on the ends of the loop and an mtz file with information for calculating a map, tell it how many residues are in the loop including the ends and how hard to try, and RESOLVE will fit the loop. It takes just a few seconds to run in most cases. If you want to do this in a fully automated way, try using the ResolveBuild and IterativeBuild wizards in PHENIX!
Automated fitting of flexible ligands to electron density maps with the resolve_ligand_fit script: RESOLVE now is capable of fitting ligands with many rotatable bonds to maps. RESOLVE starts by finding the location and orientation of the largest fixed part of the ligand, then builds all the other parts sequentially to this core. You can even give RESOLVE a list of ligands and the resolve_ligand_id script will fit each one to the map and score them all, identifying which may be correct.
Merging of NCS copies is now automatically carried out during iterative model-building and refinement with the RESOLVE_BUILD script.
New standard procedures: The best way to use SOLVE/RESOLVE now on SAD/MAD/MIR data is: (1) edit and run one of the standard SOLVE scripts, (2) follow this by editing and running the RESOLVE_BUILD script, which begins with the output of SOLVE and then does pattern-matching, fragment identification, density modification, and iterative autobuilding. The RESOLVE_BUILD script is improved from the original script in version 2.06. With fast model-building, the whole process takes only a few hours for a small protein and overnight to a few days for a moderate-sized one.
Evaluate your final model automatically: The RESOLVE_BUILD script will automatically calculate a prime-and-switch composite omit map at the very end of model-building and evaluate the model and give you a report on how well your model fits the map. You can also use this script to evaluate a model you built yourself.
Local pattern matching: RESOLVE_PATTERN can identify local patterns in your map and use them to improve your phases. Local pattern matching can make a big difference if your map is of moderate quality. This is automatically carried out using the new RESOLVE_BUILD script.
CCP4 version 4 libraries: CCP4 version 4 libraries are used in versions of SOLVE/RESOLVE starting with version 2.11. Version 2.13 will work with output from previous versions. You can compile SOLVE/RESOLVE yourself with version 5 libraries if you wish (the differences are minimal for SOLVE/RESOLVE).
Fragment identification: RESOLVE can identify the presence of fragments of structure (helices, strands) in your map and use them to improve your phases. Like local pattern matching, this can make a big difference if your map is of moderate quality. This is also automatically carried out using the new RESOLVE_BUILD script.
Superquick model-building: RESOLVE now can build your model at a rate of up to 1 residue every 2-3 seconds if you have a good map ("superquick_build"). Even the more thorough standard model-building in RESOLVE is now 3 times faster than earlier versions.
Swap-space needs for SOLVE/RESOLVE: For the standard versions, 1 GB or more of swap space is recommended (700 MB minimum). On linux machines you can now run resolve_huge, and even go as high as "isizeit = 36" if you have 4 GB of swap space. RESOLVE runs best on linux machines if they have 1 GB or more of memory
You can give SOLVE and RESOLVE all your MAD/SAD data and it will decide at what resolution the signal-to-noise is high enough to use for phasing.
SOLVE carries out all the steps of macromolecular structure determination from scaling data to calculation of an electron density map, automatically.
RESOLVE uses statistical density modification (previously called maximum-likelihood density modification) to improve electron density maps
Prime-and-switch phasing in RESOLVE removes model bias from model-phased maps. See some amazing prime-and-switch examples!
RESOLVE automatically identifies NCS in heavy-atom sites and applies it for you.
Version 2.13 contains all of earlier SOLVE and RESOLVE versions. Now you download SOLVE and RESOLVE both at once.
Your version 2 license is good for all versions 2.xx of SOLVE/RESOLVE. No need for new access codes.